
page content
Properties of metalorganic quantum spin systems and spin crossover polymers
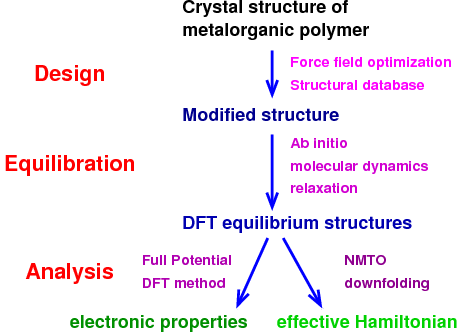
Structural design procedure for metalorganic materials
The study of metalorganic complexes has proven an innovative line of
interdisciplinary research between physics and chemistry. In the
process of the research, it turned out that in many metalorganic
systems with interesting properties, a careful preparation of
structures is a prerequisite before any further DFT analysis is
possible. Therefore, the main methodological focus of this activity
has been the development of a procedure that makes these materials
accessible to precise electronic structure calculations; the approach
can be characterized as structural design procedure for metalorganic materials.
The usefulness of this development for future studies will become
clear in the following description.
a) Design of the magnetic properties of a Cu2+ coordination polymer
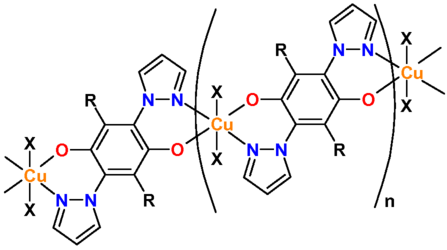
A first study addressed the question whether the magnetic properties,
namely the size of the interactions and the dimensionality of the
underlying Hamiltonian, can be designed in the computer. This idea is
based on the fact that synthetic chemistry is able to introduce small
structural modifications which are significant for the magnetic
interactions but do not change the basic crystal structure. Thus, the
design of a material would proceed by introducing feasible
substitutions or ligands to form a hypothetical structure which could
then be recommended for synthesis if the analysis proved promising.
This is a task of significant complexity because metalorganic
materials have large unit cells containing many atoms, and the precise
description of the transition metal centers translate to high demands
on the precision of the DFT calculations. Therefore, achieving our
aim required assembly of various force field and DFT methods, each of
which had a precise purpose in the procedure: The design stage in
which a hypothetical structure is constructed employs a force field
because only this computationally light weight approach allows a
global optimization, i.e. the search for the space group and lattice
parameters of the new compound. The second, equilibration stage is
performed with ab initio molecular dynamics; in this way, a
local optimization (relaxation of the fractional coordinates) of the
structures leads to the precise DFT equilibrium structure and is the
prerequisite for the subsequent analysis: On the one hand, rough
structures out of the force field relaxation often cause convergence
problems in methods like FPLAPW and LMTO, and on the other, magnetic
interaction pathways are very susceptible to even small changes in
bond lengths and angles, and thus the analysis would be meaningless if
it is not conducted on precise equilibrium structures. The third,
analysis stage then involves calculation of the band structures and
extraction of the underlying Hamiltonian by NMTO downfolding and by
calculation of total energy differences between different spin
configurations. The material that was chosen for testing the outlined
approach is a coordination polymer containing Cu2+ centers and
forms an antiferromagnetically coupled S = 1/2 Heisenberg chain. The
crystal structure of this recently synthesized material [1] has
been resolved by X-ray diffraction, and the exchange coupling of J = 20K was determined from the measured susceptibility. It turned out that
it is possible to significantly vary the band width of the low energy
bands close to the Fermi level and thus modify the exchange interaction
strength between minus 90% and plus
25% via side chains to the polymer backbone or ligands. The
modifications also proved suitable to change the dimensionality from
purely one-dimensional towards more two- or even three-dimensional.
The results of this study are published in
Refs. [JSV+07,SJR+07].
b) Spin dimer system C36H48Cu2F6N8O12S2 (TK91)
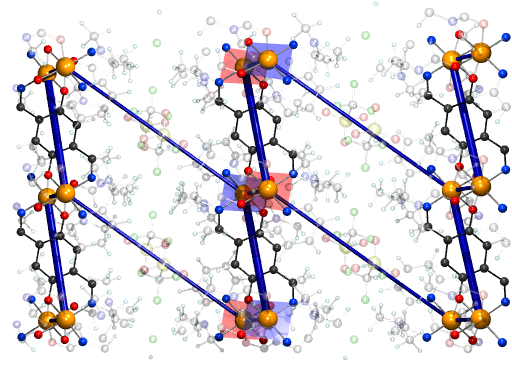
In the recently synthesized material TK91, two Cu2+ ions
have been bridged via hydroquinone linkers [2]. The resulting
Cu2+ dimers are arranged in the crystal in staggered
chains. In a magnetic field, this spin dimer system exhibits an exotic
magnetic state (probably a Kosterlitz-Thouless condensation of
magnetic excitations). With the help of NMTO downfolding, we find that
besides the dominant coupling in the Cu2+ dimer, there are,
out of a large number of possible interaction pathways, only two
further important interactions between Cu2+ centers. This
allows the elucidation of the connectivity of the underlying model
Hamiltonian which corresponds to a 2D network. Quantum Monte Carlo
calculations with the model obtained in this way yields excellent
agreement with the measured data. A publication is in preparation.
The method of designing metalorganic materials with certain magnetic
properties developed and tested on the Cu2+ polymer will
become important when it is applied to materials that show important
effects like TK91. Here, tuning of the material can yield insight
into complex many body effects.
c) Microscopic investigation of a spin crossover coordination complex
In a second study, we have employed the method outlined under a) for
the study of the microscopic origins of spin crossover. Spin crossover
is an important phenomenon in which transition metal ions can be
changed from a low spin to a high spin state by a change in temperature
or pressure. This phenomenon has up to now been described by
phenomenological models that assumed the mechanism of spin crossover
to be elastic in origin; the purpose of the present investigation was
to make progress towards a microscopic understanding of the
phenomenon. Spin crossover is especially interesting in low
dimensional systems because this makes the effect cooperative,
leading to a large hysteresis in the transition temperatures.
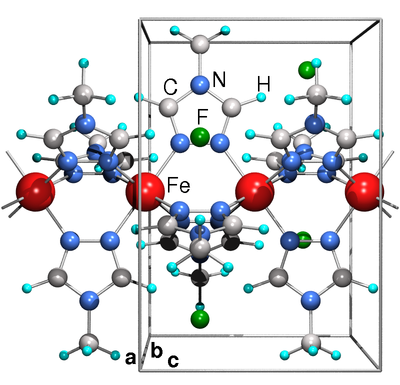
An important family of spin crossover polymers are Fe2+ triazole
systems. The difficulty for a microscopic study is the fact that these
polymers form only nanocrystalline powders and thus their structure
cannot be resolved with certainty. Besides, even candidates for the
crystal structure contain so many atoms in the unit cell that the
computational effort would be prohibitive. Therefore our study
proceeds in two steps: in a first step, we use force field global
optimization to construct a simplified model system that contains the
crucial Fe2+ triazole backbone but has simplified counterions and
side chains. In the second step, based on the fact that the spin state
of iron depends on the bond lengths in the FeN6 octahedron, we
construct a series of structures with different sizes of the FeN6
octahedron and relax them first with force field, then with Car
Parrinello molecular dynamics methods. The structures obtained in this
way show the transition from low spin to high spin in a straight
forward way in spin resolved DFT. Furthermore, with the help of NMTO
downfolding as well as total energy differences we can determine the
strength of the magnetic exchange between Fe2+ centers in the
polymer. We find that this exchange interaction is significant, making
magnetic interactions equally important as elastic interactions for
explaining the cooperative effect of the large hysteresis between high
spin and low spin states. This study is published in
Refs. [JSV+07,JSR+07].